Plug & Play Directed Evolution of Proteins with Gradient-based Discrete MCMC
Patrick Emami, Aidan Perreault, Jeff Law, David Biagioni, Peter C. St. John. Machine Learning: Science and Technology, 2023. Presented at NeurIPS’22 Workshop on Machine Learning in Structural Biology.
[DOI] [arxiv] [Paper code] [EvoProtGrad library]
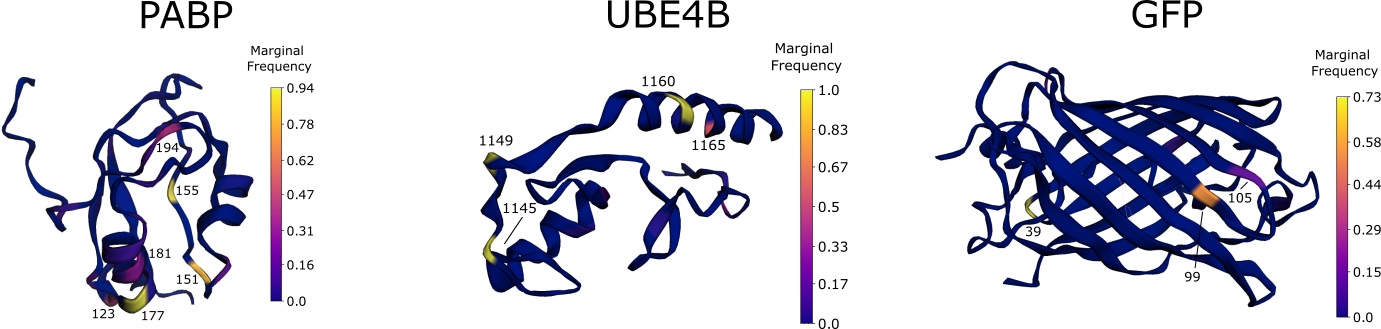
The marginal frequency of each sequence position visualized on the 3D structure of the wild type is the fraction of protein variants in the sampled variant pool that have a mutation at that sequence position. For example, nearly 100% of discovered UBE4B variants have a mutation at position 1145.
Announcing EvoProtGrad
7/21/23: We have released a Python package accompanying this work with HuggingFace integration for supporting a wide range of protein language models. Check it out on Github.
pip install evo_prot_grad
Quick demo
Evolve the avGFP protein with Rostlab/prot_bert.
import evo_prot_grad
prot_bert_expert = evo_prot_grad.get_expert('bert', temperature = 1.0)
variants, scores = evo_prot_grad.DirectedEvolution(
wt_fasta = 'test/gfp.fasta', # path to wild type fasta file
output = 'best', # return best, last, all variants
experts = [prot_bert_expert], # list of experts to compose
parallel_chains = 1, # number of parallel chains to run
n_steps = 20, # number of MCMC steps per chain
max_mutations = 10, # maximum number of mutations per variant
verbose = True # print debug info to command line
)()
Summary
Machine-learning-based directed evolution can be used to engineer new proteins that have improved productivity or catalyze new reactions. Basically, starting from a known protein sequence, directed evolution proceeds by identifying candidate mutations, experimentally verifying them, and then using the gained information to search for better mutations. The intention is for this iterative process to resemble natural evolution. One way ML can help streamline this design loop is by optimizing the candidate indentification step.
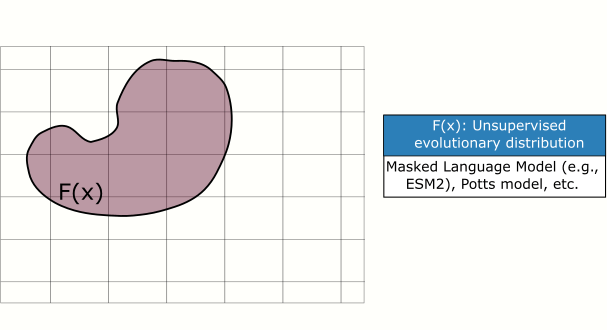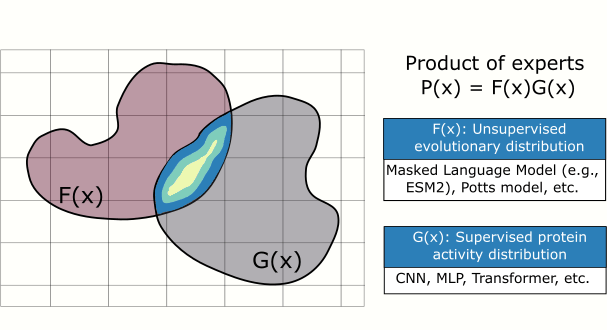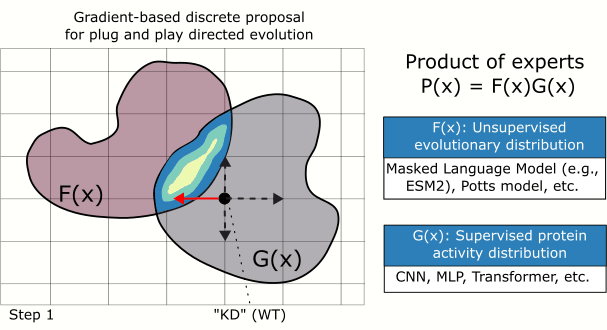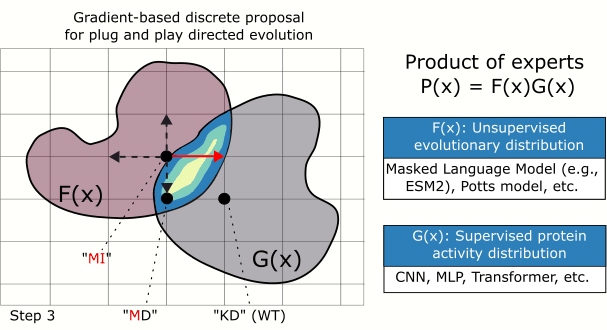
In this paper, we introduce a plug and play protein sampler for this step. The sampler mixes and matches unsupervised evolutionary sequence models (e.g., protein language models) with supervised sequence models that map sequence to function, without needing any model re-training or fine-tuning. Sampling is performed in sequence space to keep things as ‘‘black-box’’ as possible. Plug and play samplers have been successful in other domains, such as for controlled image and text generation.
Searching through protein sequence space efficiently, which is high-dimensional and discrete, is notoriously difficult. We address this in our MCMC-based sampler by using the gradient of the (differentiable) target function to bias a mutation proposal distribution towards favorable mutations. We show that this approach outperforms classic random search.
Impact: Improving the quality of candidate protein variants reduces the time spent conducting costly experiments in the wet lab and accelerates the engineering of new proteins.
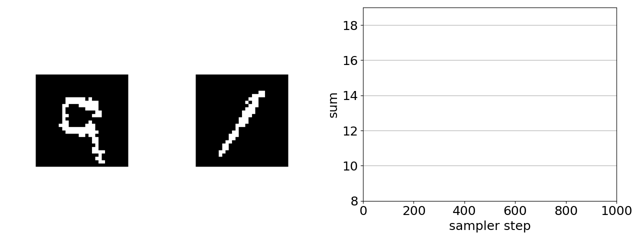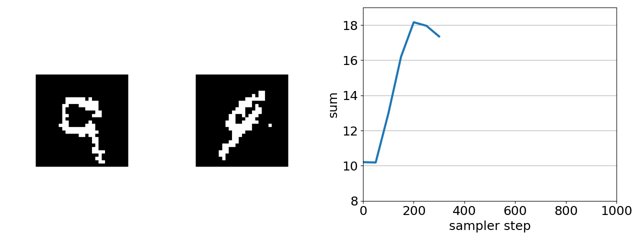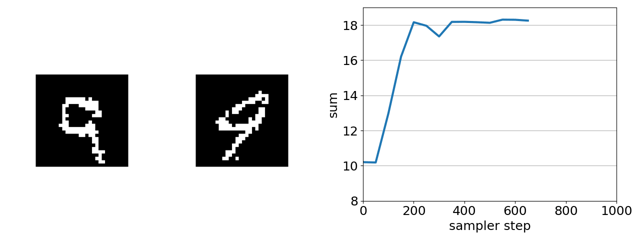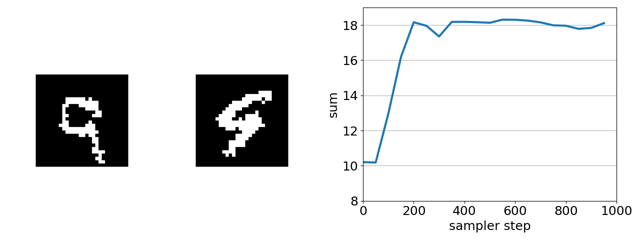
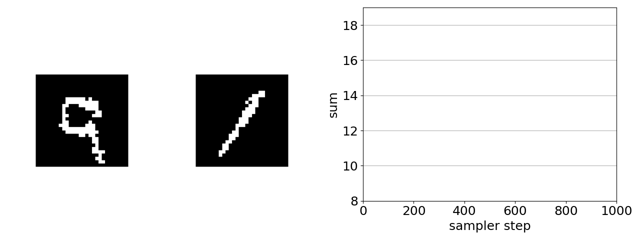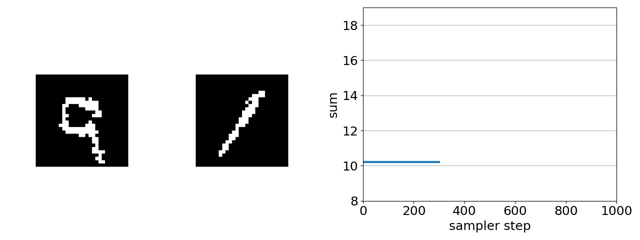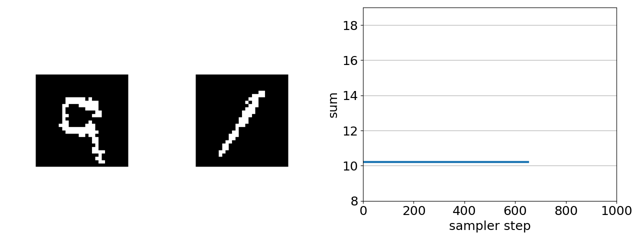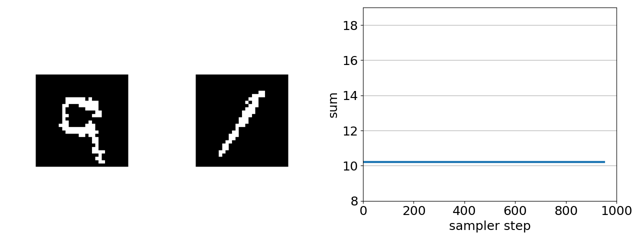
Checking intuitions with a toy binary MNIST experiment
The task is to evolve the MNIST digit on the right, initially a 1, to maximize the sum of the two digits solely by flipping binary pixels. Since the digit on the left is a 9, the initial sum of the digits is 10. The maximum is 18 (9 + 9).
Here’s our sampler applied to a product of experts distribution that combines an unsupervised energy based model with a supervised ensemble of ConvNets trained to regress the sum.
If we remove the EBM from the product of experts, the search gets trapped in adversarial optima—white-noise images. This is because, in this case, gradient-based discrete MCMC is only using the gradient of the ConvNet ensemble to steer search. The EBM acts as a soft constraint that keeps search away from such optima.
If we replace gradient-based discrete MCMC with random-search-based simulated annealing, nearly every randomly sampled mutation is rejected as it does not increase the predicted sum.
In-silico directed evolution experiments
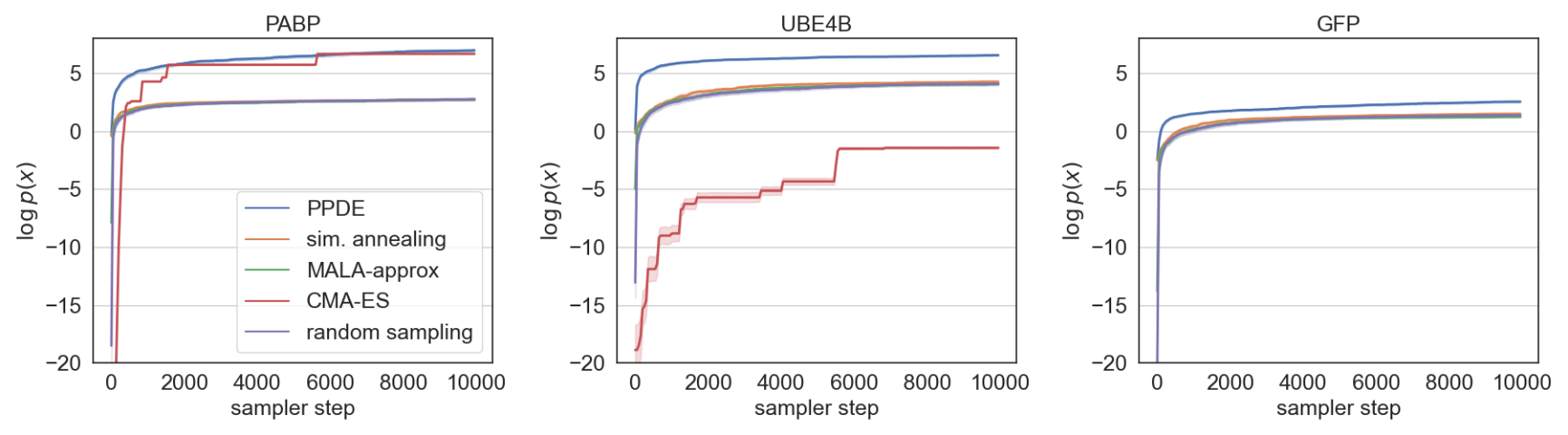
We conduct in-silico directed evolution experiments on 3 proteins with partially-characterized wide fitness landscapes (between 2-15 mutations from wild type): PABP_YEAST, UBE4B_MOUSE, and GFP.
Compared to random-walk-based black-box samplers (simulated annealing, Metropolis-adjusted Langevin Algorithm (MALA)), random sampling, and an evolutionary method (CMA-ES), our sampler explores sequence space most efficiently (higher $\log p(x)$ is better).
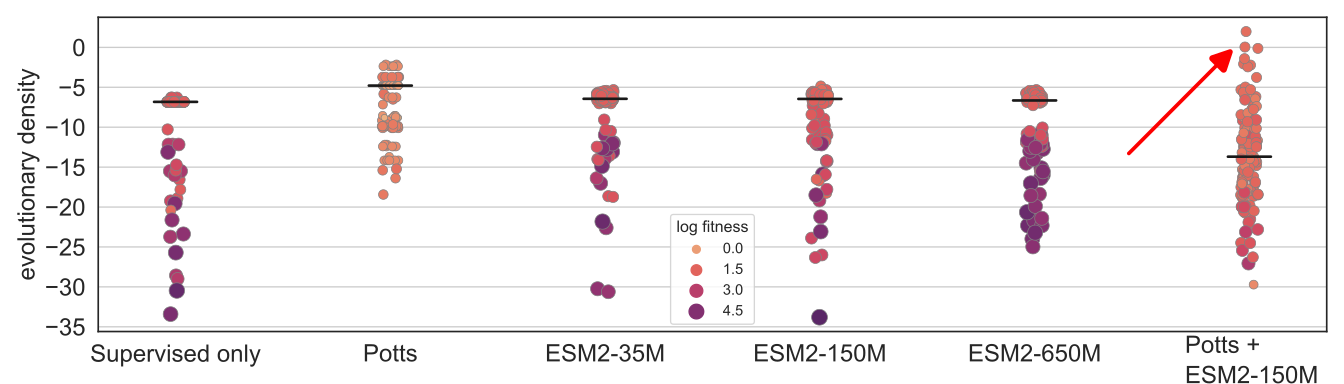
We show that we can plug in different protein language models: ESM2-35M, ESM2-150M, and ESM2-650M. We also combine ESM2-150M with a Potts model, which surprisingly achieved the best performance (red arrow).
See the paper and code for more details and results.